Généralités
L’hypothyroïdie congénitale (HC) est une maladie liée à une sécrétion insuffisante d’hormone thyroïdienne par la glande thyroïde. Le terme « congénital » signifie que cette anomalie est présente dès la naissance, même si elle ne donne pas toujours de symptômes immédiats. C’est la maladie endocrinienne la plus fréquente du nouveau-né puisqu’elle atteint 1/3500 à 1/4000 nouveau-nés. En l’absence de traitement précoce, l’HC entraîne un retard sévère du développement psychomoteur et de la croissance. Grâce au dépistage néonatal systématique, mis en place depuis 1978, l’enfant atteint bénéficie précocement d’un traitement hormonal substitutif (l’hormone thyroïdienne qui lui manque), ce qui lui permet de se développer normalement.
Qu’est-ce que la glande thyroïde?
La thyroïde est une petite glande située à la partie basse du cou (figure 1).
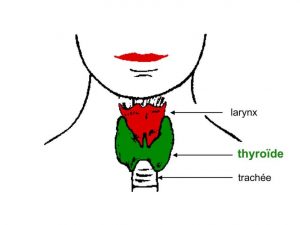
Figure 1: Localisation de la glande thyroïde dans le cou
Elle produit des messagers, les hormones thyroïdiennes, qui sont déversées dans le sang et agissent ainsi dans tout l’organisme: la « T3 » (triiodothyronine) et la « T4 » (thyroxine). Ces hormones thyroïdiennes jouent un rôle capital dans la croissance staturale et le développement du cerveau. Leur production est régulée par une autre hormone, la « TSH » (Thyroid Stimulating Hormone) qui est, elle, sécrétée par la glande hypophyse (figure 2).
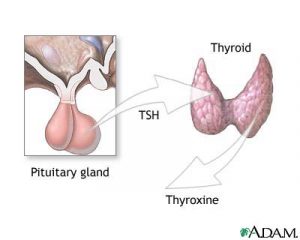
Figure 2 : Régulation de la sécrétion des hormones thyroïdiennes par la TSH
Comment est fait le diagnostic?
Le dépistage est systématique à la naissance chez tous les nouveau-nés en France à partir de quelques gouttes de sang prélevées par une simple piqûre au talon au 3ème jour de vie. Il repose sur la mise en évidence d’un taux sanguin élevé de TSH (en cas d’hypothyroïdie, l’organisme essaie en vain de forcer la production des hormones thyroïdiennes en augmentant le taux de TSH dans le sang).
Le diagnostic d’HC est confirmé par un nouveau prélèvement sanguin qui montre un taux élevé de TSH et un taux très bas de T4. Des examens complémentaires sont réalisés dès le diagnostic confirmé pour déterminer la cause da la maladie :
- La scintigraphie thyroïdienne permet d’observer le fonctionnement de la glande et la fabrication des hormones. C’est une technique d’imagerie qui utilise un produit radioactif inoffensif (iode ou technetium) qui se fixe sur la thyroïde. Elle permet la plupart du temps de déterminer s’il y a une anomalie de la synthèse fabrication hormonale et de visualiser la thyroïde si elle n’est pas en position normale.
- L‘échographie thyroïdienne permet aussi de localiser la thyroïde et donc de mettre en évidence une absence, un développement insuffisant ou une situation adéquate de la glande.
Quelles sont les manifestations de l’hypothyroïdie congénitale?
A la naissance, l’enfant ayant une HC ne présente aucun signe visible. Grâce, au dépistage néonatal systématique, la maladie est en générale diagnostiquée avant que les symptômes d’hypothyroïdie n’apparaissent. Certains signes mineurs peuvent cependant faire anticiper le diagnostic des médecins avertis : jaunisse prolongée, difficultés à téter, enfant trop « endormi », peau marbrée, fontanelles larges…
Quelle est la cause de l’hypothyroïdie congénitale?
Les causes d’HC sont diverses :
- Dans 85% des cas, il s’agit d’une malformation ou anomalie de développement de la glande thyroïde. On distingue les cas où la glande est absente (athyréose): 25% des HC; les cas où la glande est en situation anormale entre la base de langue et la base du cou (ectopie): 50% des HC; enfin, les cas où la glande a un développement insuffisant (thyroïde hypoplasique, hémithyroïde…) : 5 à 10% des cas. Dans ces cas-là, la thyroïde anormale produit des hormones en quantité insuffisante pour couvrir les besoins de l’enfant. Ces formes sont le plus souvent sporadiques c’est-à-dire que le risque d’avoir un nouvel enfant atteint est minime voire nul. Dans 2% des cas cependant, il s’agit de formes familiales avec donc un risque de récidive dans la famille.
- Dans 15% des cas, la thyroïde est bien développée, bien située, mais ne fabrique pas normalement les hormones thyroïdiennes. On parle de trouble de l’hormonosynthèse thyroïdienne. L’anomalie peut toucher la synthèse fabrication, , ou la sécrétion libération. Ces formes sont transmises de manière récessive, ce qui signifie que les parents ne sont pas malades, mais qu’il y a, à chaque grossesse, un risque sur 4 d’avoir un enfant atteint d’HC .
- Beaucoup plus rarement, la sécrétion insuffisante de la thyroxine est la conséquence d’une anomalie de l’hypophyse qui sécrète en quantité insuffisante de la TSH (figure 2). Ces formes échappent au dépistage néonatal.
- Il existe enfin des hypothyroïdies transitoires : soit liées au passage d’anticorps maternels vers le fœtus au travers du placenta, ces anticorps empêchant la TSH de commander la sécrétion d’hormones thyroïdiennes; soit liées à un traitement pris par la mère au cours de la grossesse (antithyroïdiens, produits iodés).
Quels sont les gènes impliqués dans les formes familiales?
Les gènes candidats associés à l’HC, peuvent être divisés en 2 groupes principaux :
- Ceux responsables d’une anomalie de développement de la glande thyroïde:
- Formes isolées: mutation du gène du récepteur de la TSH
- Formes syndromiques (association à d’autres anomalies): Mutation des gènes codant pour la protéine Gsα, les facteurs de transcription impliqués dans le développement de la thyroïde (TTF-1, TTF-2, Pax-8) .
- Ceux responsables des troubles de l’hormonosynthèse thyroïdienne: mutation des gènes codant pour la thyroperoxydase, la thyroglobuline, le transporteur d’iode, la pendrine, la thyroïde oxydase 2.
En quoi consiste le traitement?
Le traitement consiste à donner par voie orale, tous les jours, sous forme de gouttes, l’hormone thyroïdienne de synthèse (L-thyroxine®) qui manque à l’enfant. Le traitement est débuté le plus tôt possible (généralement entre le 8ème et le 10ème jour de vie) et doit être poursuivi toute la vie. La posologie initiale recommandée a augmenté au cours des dernières décennies pour atteindre désormais 10 à 15 µg/kg/jour. La prescription initiale est ensuite ajustée individuellement par l’endocrinologue en fonction des contrôles sanguins de la TSH et de la T4 ; ces contrôles fréquents au début (mensuel au minimum) s’espacent ensuite (semestriel au minimum).
Quels sont les bénéfices attendus?
Le dépistage néonatal systématique a complètement transformé le pronostic mental des HC. Actuellement, avec un recul d’une trentaine d’années, le développement psychomoteur des hypothyroïdies dépistées est considéré comme normal pour l’ensemble des enfants détectés. Cependant, plusieurs études font état d’un pourcentage (évalué à 10%) d’hypothyroïdies qualifiées de sévères, dont le développement mental est légèrement inférieur à celui de la population générale. Trois facteurs prédictifs du devenir mental ont récemment été mis en évidence :
- la précocité d’institution du traitement
- une dose thérapeutique initiale forte de 10 à 15 µg/kg/jour permet de normaliser le développement psychomoteur de ces formes dites sévères
- l’observance du traitement toute au long de la vie est essentielle.
Nous pouvons donc considérer qu’actuellement, les enfants se développent normalement et mènent une vie normale, à condition de prendre leur traitement à vie.
Une forme particulière: l’hypothyroïdie congénitale par mutation inactivatrice du récepteur de la TSH
De rares cas d’hypothyroïdie congénitale sont causés par des mutations du gène codant pour le récepteur de la TSH. Le récepteur de la TSH est situé à la surface des cellules thyroïdiennes. La fixation de la TSH (hormone secrétée par une petite glande située à la base du cerveau: l’hypophyse) sur ces récepteurs active la multiplication et la différenciation des cellules thyroïdiennes ainsi que la sécrétion des hormones thyroïdiennes T3 et T4 par les cellules thyroïdiennes (figure 2). Le récepteur de la TSH joue donc un rôle essentiel dans le développement et le fonctionnement de la glande thyroïde. Son gène est localisé sur le chromosome 14 en 14q31. Les patients atteints sont soit homozygotes pour la mutation (une mutation identique sur chaque chromosome) soit hétérozygotes composites (double hétérozygotes, une mutation différente sur chaque chromosome. Dans ce cas, le récepteur est inactivé; la TSH ne peut plus activer son récepteur: il se crée une « résistance à la TSH ». Ces cas d’hypothyroïdies congénitales se présentent sous la forme d’hypoplasie de la glande thyroïde avec biologiquement une élévation très importante du taux de TSH et une baisse des hormones thyroïdiennes. Le traitement est classiquement une supplémentation en L-thyroxine à vie.
Docteur Natacha BOUHOURS-NOUET